Ja, das Molekül sollte eine Chiralität unterhalb der Raumtemperatur aufweisen.
In der optimierten 1 sup> -Molekülkonfiguration kann das Molekül seinem Spiegelbild nicht überlagert werden. Dies liegt an der Rotation der Nitrogruppen außerhalb der Ebene.
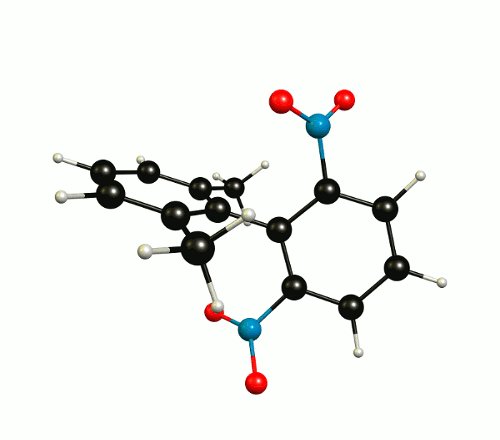
Die thermodynamisch günstigste Struktur gehört zur Klasse $ C_2 $ span> -Symmetrie und hat keine Spiegelebene (siehe oben).
Im Gegensatz zu den zuvor vorgestellten Argumenten ist eine Analyse der Lewis-Struktur irreführend, da sie massiv falsch einschätzt der Platzbedarf der Nitroeinheiten. Ein Merkmal, das bereits in 2,4,6-Trinitrotoluol vorhanden ist. 2 sup> Aus der Zusammenfassung von Clarkson et al. : 3 sup>
Es wurden zwei echte Energieminimumstrukturen gefunden. In beiden Strukturen ist die 4-Nitrogruppe planar zum Phenylring, während die 2,6-Nitrogruppen aufgrund der sterischen Wechselwirkung mit der Methylgruppe leicht außerhalb der Ebene des Phenylrings liegen.
Sie beschreiben später:
Die beiden stabilen Molekülstrukturen von TNT, Abb. 1, hängen durch interne Rotationen der 2- und 6-Nitrogruppen und der Methylgruppe zusammen. [... ] Struktur A ist die stabilere Geometrie, obwohl der Energieunterschied gering ist ( $ 0.650 ~ \ pu {kcal mol ^ −1} $ span> bei B3LYP / 6-311 + G **, Tabelle 4). Struktur A zeigt nahezu ideale $ C_ \ mathrm {s} $ span> -Symmetrie mit einem der Methylwasserstoffatome senkrecht zum Phenylring in der Ebene von $ \ sigma_ \ mathrm {h} $ span>. Die 2,6-Nitrogruppen von A sind nicht planar und werden in dieselbe Fläche des Phenylrings gedreht, wodurch die Anzahl der Van-der-Waals-Wechselwirkungen mit der Methylgruppe maximiert wird.
Die 2,6-Phenyleinheit ist jedoch viel größer als eine Methylgruppe, weshalb die Symmetrie der Nitrogruppen gebrochen ist. Das Erzwingen dieser $ C_ \ mathrm {s} $ span> -Symmetrie führt zu einem Sattelpunkt erster Ordnung (Übergangszustand) auf der potenziellen Energieoberfläche, der nur etwa $ 1.6 ~ \ pu {kJ mol ^ -1} $ span> hat eine höhere Energie als die Mindeststruktur.
Bei Raumtemperatur ist es wahrscheinlich, dass die gegenseitige Umwandlung zwischen den Enantiomeren dadurch erfolgt Struktur. Ich habe nicht überprüft, ob dieser Zustand tatsächlich über eine intrinsische Reaktionskoordinatenberechnung verbunden ist, aber aus der visuellen Untersuchung des imaginären Modus scheint dies sehr wahrscheinlich zu sein.
Wenn Sie das System ausreichend kühlen, sollten Sie in der Lage sein, zwei Enantiomere zu identifizieren. Bei Raumtemperatur sollte die gegenseitige Umwandlung jedoch zu schnell sein, um beobachtet zu werden.
Es wäre gut zu wissen, welches Buch Sie verwendet haben und in welchem Kontext die Erklärung abgegeben wurde. Aus rein theoretischer Sicht würde ich zustimmen, dass das Molekül chiral ist. Aus praktischer Sicht würde ich diese Eigenschaft nicht bewerten, da sie wahrscheinlich zu schwer zu messen ist.
Der vorgeschlagene $ C_ \ mathrm {2v } $ span> -Strukturen sind je nach Rotation der Methylgruppen entweder Sattelpunkte zweiter oder vierter Ordnung. Diese Strukturen haben mindestens $ 15 ~ \ pu {kJ mol ^ -1} $ span> eine höhere Energie als die ersteren und keinen brauchbaren Weg für die gegenseitige Umwandlung.

Fußnoten
- Alle Strukturen wurden auf der theoretischen Ebene DF-M06L / def2-SVP mit Gauß optimiert 09 Rev. D01. Stationäre Punkte wurden durch Berechnung der Schwingungsfrequenzen auf derselben theoretischen Ebene charakterisiert.
- W. Robert Carper, Larry P. Davis, Michael W. Extine, J. Phys. Chem. 1982, 86 (4), 459–462.
- John Clarkson, W. Ewen Smith, David N. Batchelder, D. Alastair Smith, Alison M. Coats, J. Mol. Struc. 2003, 648 (3), 203-214.