Es gibt viele gute Übersichtsartikel, aber ich mag das frei verfügbare "The ABC of DFT" von Kieron Burke (und Freunden) und einen kürzlich erschienenen Übersichtsartikel versucht, die verständliche Sprache beizubehalten.
Das Problem ist die Behandlung der Coulomb-Interaktionen. Herkömmliche DFT ist nicht "asymptotisch korrekt". Das heißt, auf große Entfernung wird das 1 / r-Verhalten nicht erfüllt, und Dispersionswechselwirkungen (z. B. Van-der-Waals-Kräfte) werden ebenfalls nicht richtig gehandhabt.
Zum Beispiel in Neonatomen (aus ABC von DFT von Burke): 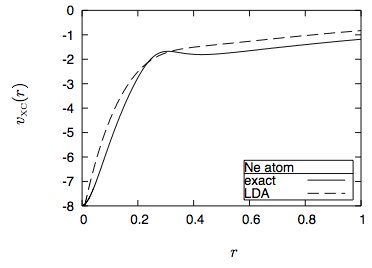
Das Problem ist natürlich, dass die meisten DFT-Methoden von der Local Density Approximation (LDA) ausgehen, bei der nur die Elektronendichte bei einem bestimmten ( lokaler) Punkt. LDA gibt an, dass das Fernverhalten exponentiell anstelle von 1 / r abfällt:
$$ v_ {xc} ^ {\ mathrm {LDA}} (\ mathbf {r}) = \ frac {\ Delta E ^ {\ mathrm {LDA}}} {\ delta \ rho (\ mathbf {r})} = \ epsilon_ {xc} (\ rho (\ mathbf {r})) + \ rho (\ mathbf {r}) ) \ frac {\ partielle \ epsilon_ {xc} (\ rho (\ mathbf {r}))} {\ partielle \ rho (\ mathbf {r})} \ $$
Also einige neuere Bemühungen haben "bereichsgetrennte" Funktionen verwendet, z. B.
$$ \ frac {1} {r} = \ frac {1-g (r)} {r} + \ frac {g (r)} {r} $$
Der erste Term (mit kurzer Reichweite) kann durch die generalisierte Gradientennäherung (GGA) behandelt werden, die von praktisch allen modernen Funktionalen verwendet wird. Der zweite Term wird normalerweise von Hartree-Fock behandelt (dh exakter Austausch), und es gibt eine Funktion g (r), die sich reibungslos zwischen den beiden skalieren lässt.
Ich denke, moderne, durch Entfernungen getrennte Hybridfunktionen sind ziemlich gut Die Auswahl einer Funktion hängt jedoch ein wenig von der Eigenschaft ab, die Sie vorhersagen möchten. Thermodynamik? Anregungsenergien? Oxidations- / Reduktionspotentiale aus orbitalen Eigenwerten? usw.