Zusätzlich zu @ Nanoputians hervorragender Beschreibung konstruktiver und destruktiver Interferenzen bei der Bildung von MOs möchte ich eine mathematischere Erklärung dafür liefern, warum die Phase der Wellenfunktion keine Rolle spielt.
Finden der Wellenfunktion
Die zeitunabhängige Schrödinger-Gleichung lautet in einer Dimension:
$$ \ hat {H} \ psi (x) = E \ psi (x) $ $
Es kann gezeigt werden, dass, wenn eine Wellenfunktion $ \ psi = \ psi (x) $ die obige Gleichung erfüllt, die Wellenfunktion $ k \ psi $ (mit $ k \ in \ mathbb {C}) erfüllt. $) erfüllt auch die obige Gleichung mit dem gleichen Energieeigenwert $ E $. Dies liegt an der Linearität des Hamilton-Operators:
$$ \ begin {align} \ hat {H} (k \ psi) & = k (\ hat {H}) \ psi) \\ & = k (E \ psi) \\ & = E (k \ psi) \ end {align} $$
Es gibt mehrere Bedingungen, die eine Wellenfunktion erfüllen muss es muss physikalisch realisierbar sein, dh es soll ein "reales" physikalisches Teilchen darstellen. In dieser Diskussion ist die relevante Bedingung, dass die Wellenfunktion quadratintegrierbar (oder normalisierbar ) sein muss. In mathematischen Begriffen:
$$ \ langle \ psi \ lvert \ psi \ rangle = \ int _ {- \ infty} ^ {\ infty} \! \ Lvert \ psi \ rvert ^ 2 \, \ mathrm {d} x < \ infty $$
Dies bedeutet, dass in \ mathbb {C} $ eine Konstante $ N \ existieren muss, sodass $ N \ psi $ normalisiert em ist >:
$$ \ int _ {- \ infty} ^ {\ infty} \! \ lvert N \ psi \ rvert ^ 2 \, \ mathrm {d} x = \ lvert N \ rvert ^ 2 \! \! \ int _ {- \ infty} ^ {\ infty} \! \ lvert \ psi \ rvert ^ 2 \, \ mathrm {d} x = 1 $$
Von diesem Punkt an Wir gehen davon aus, dass wir bereits die geeignete Normalisierungskonstante gefunden haben, so dass die Wellenfunktion $ \ psi $ bereits normalisiert ist. Mit anderen Worten, nehmen wir an, $ \ langle \ psi \ lvert \ psi \ rangle = 1 $, weil wir können. Betrachten wir nun die Wellenfunktion $ - \ psi $, die $ N \ psi $ mit $ N = -1 $ entspricht. Ist diese neue Wellenfunktion normalisiert?
$$ \ begin {align}
\ int _ {- \ infty} ^ {\ infty} \! \ lvert - \ psi \ rvert ^ 2 \, \ mathrm {d} x & = \ lvert -1 \ rvert ^ 2 \! \! \ int _ {- \ infty} ^ {\ infty} \! \ lvert \ psi \ rvert ^ 2 \, \ mathrm {d} x \\ & = \ int _ {- \ infty} ^ {\ infty} \! \ lvert \ psi \ rvert ^ 2 \, \ mathrm {d} x \\ & = 1 \ end {align} $$
Natürlich ist es das. Was ich bisher geschrieben habe, lautet also im Grunde: Wenn $ \ psi $ eine normalisierte Lösung für die Schrödinger-Gleichung ist, ist dies auch $ - \ psi $ .
Tatsächlich Sie könnten noch einen Schritt weiter gehen. Mit genau der gleichen Arbeitsweise wie oben könnten Sie zeigen, dass, wenn $ \ psi $ eine normalisierte Lösung der Schrödinger-Gleichung ist, die Wellenfunktion $ (a + ib) \ psi $ auch eine wäre, solange $ a ^ 2 + b ^ 2 = 1 $. (Wenn Sie Exponentiale mögen, entspricht dies $ a + ib = e ^ {i \ theta} $.) Ich habe diese Idee in diesem Diagramm veranschaulicht:
$ \ qquad \ qquad \ qquad \ qquad \ qquad \ qquad $ 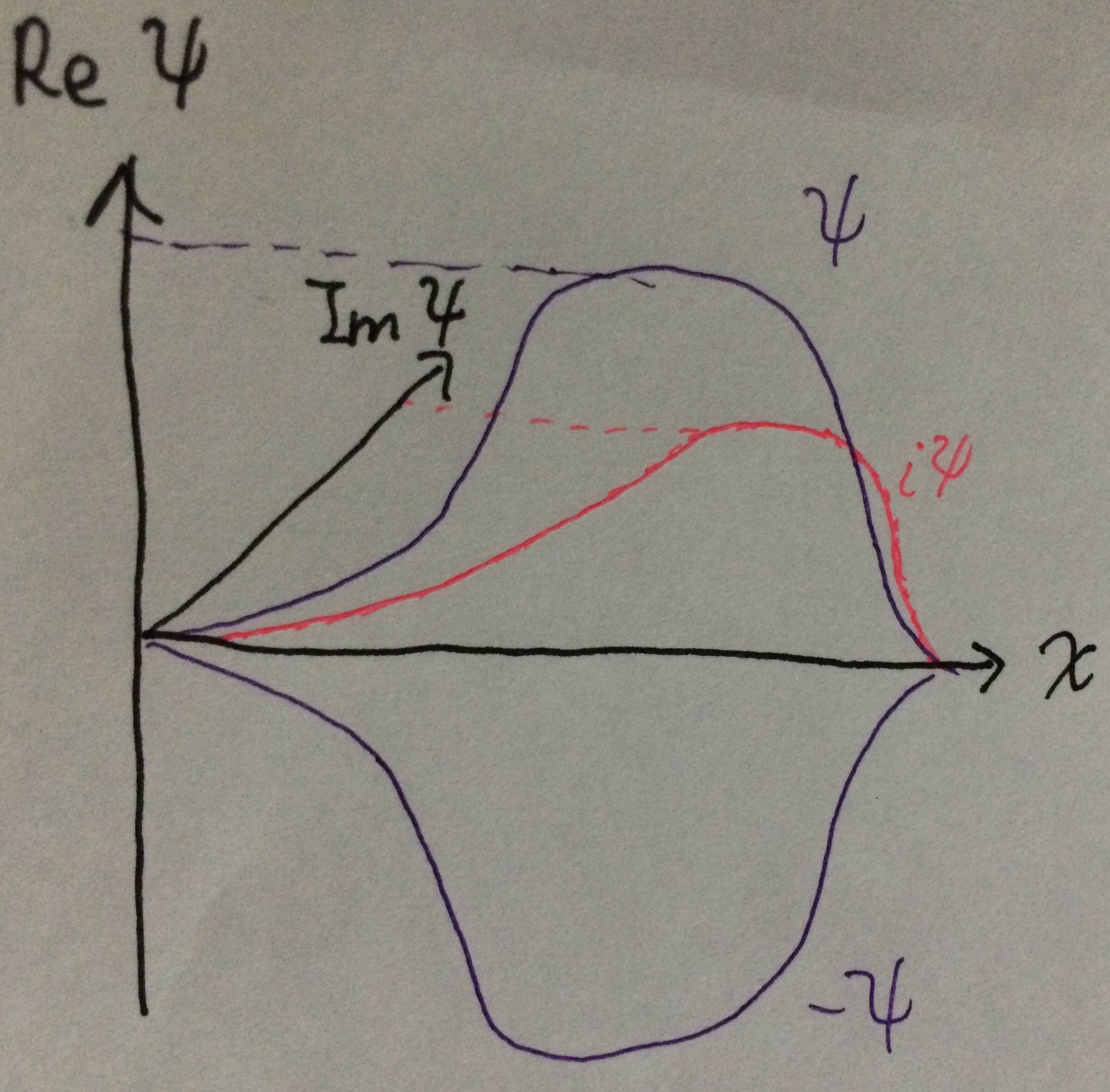
Wenn $ \ psi $ eine eindimensionale Wellenfunktion mit realem Wert ist, können Sie sie in einem Diagramm gegen $ x $ darstellen. Die Wellenfunktion $ i \ psi $ hätte dann genau die gleiche Form und würde nur aus der Papierebene herauskommen ($ \ theta = 90 ^ \ circ $). Sie könnten die Wellenfunktion $ (1 + i) \ psi / \ sqrt {2} $ haben. Es würde um $ \ theta = 45 ^ \ circ $ nach außen aus der Papierebene zeigen, genau auf halber Strecke zwischen $ \ psi $ und $ i \ psi $, aber genau in der gleichen Form. Die Physik weiß jedoch nicht, wo sich die Ebene Ihres Papiers befindet, daher sind alle diese Wellenfunktionen gleichermaßen zulässig. Aus Sicht des Systems sind sie alle dasselbe.
Verwenden der Wellenfunktion
"Aber warten Sie! Wenn die Wellenfunktion negativ ist, was? über die Werte von Impuls, Position und Energie, die Sie berechnen? Werden sie negativ? "
" Gute Frage, ich selbst! "
Nun, für den Anfang ist eine Sache, für die Sie die Wellenfunktion verwenden, die Wahrscheinlichkeitsdichte $ P (x) $ zu finden. Nach Max Borns Interpretation der Wellenfunktion ist dies gegeben durch $ P (x) = \ lvert \ psi \ rvert ^ 2 $. Angenommen, die durch die negative Wellenfunktion $ - \ psi $ beschriebene Wahrscheinlichkeitsdichte ist eine andere Funktion von $ x $, die als $ Q (x) $ bezeichnet wird:
$$ \ begin {align} Q (x ) = \ lvert - \ psi \ rvert ^ 2 & = \ lvert -1 \ rvert ^ 2 \ lvert \ psi \ rvert ^ 2 \\ & = \ lvert \ psi \ rvert ^ 2 \\ & = P (x) \ end {align} $$
Die durch die negative Wellenfunktion beschriebene Wahrscheinlichkeitsdichte ist also genau gleich. Tatsächlich ist die durch $ i \ psi $ beschriebene Wahrscheinlichkeitsdichte ebenfalls genau dieselbe.
Lassen Sie uns nun über Observable sprechen, wie z. B. Position $ x $, Momentum $ p $ und Energie $ E $. Jedes Observable hat einen entsprechenden Operator: $ \ hat {x} $, $ \ hat {p} $ und $ \ hat {H} $ (der Hamiltonianer hat einen speziellen Buchstaben, weil er nach William Hamilton benannt ist) a>). Mit diesen Operatoren berechnen Sie den Mittelwert des Observablen. Ich werde ein Beispiel bezüglich der Dynamik geben. Wenn Sie den mittleren Impuls mit der Bezeichnung $ \ langle p \ rangle $ ermitteln möchten, gehen Sie wie folgt vor:
$$ \ begin {align} \ langle p \ rangle & = \ langle \ psi \ lvert \ hat {p} \ rvert \ psi \ rangle \\ & = \ int _ {- \ infty} ^ \ infty \! \ psi ^ * \ hat {p} \ psi \, \ mathrm {d} x \ end { align} $$
Ich werde den Wert dieses Integrals $ p_1 $ aufrufen. Lassen Sie uns jetzt das Gleiche tun. Nehmen wir an, dass der mittlere Impuls für die negative Wellenfunktion nicht unbedingt der gleiche Wert ist. Nennen wir den neuen mittleren Impuls etwas anderes, wie $ p_2 $.
Bevor wir fortfahren, werde ich feststellen, dass der Impulsoperator $ \ hat {p} = -i \ hbar \ frac {\ mathrm {d}} {\ mathrm {d} x} $ ebenfalls linear ist . Wenn Sie daran zweifeln, können Sie es anhand der Definition der Linearität im allerersten Link testen, den ich gepostet habe. Tatsächlich sind alle quantenmechanischen Operatoren, die Observablen entsprechen, linear. Daher $ \ hat {p} (- \ psi) = - \ hat {p} \ psi $ und so:
$$ \ begin {align} p_2 & = \ langle - \ psi \ lvert \ hat {p} \ lvert- \ psi \ rangle \\ & = \ int _ {- \ infty} ^ \ infty \! (- \ psi) ^ * \ hat {p} (- \ psi) \, \ mathrm {d} x \\ & = (-1) ^ 2 \! \! \ int _ {- \ infty} ^ \ infty \ ! \ psi ^ * \ hat {p} \ psi \, \ mathrm {d} x \\ & = \ int _ {- \ infty} ^ \ infty \! \ psi ^ * \ hat {p} \ psi \, \ mathrm {d} x \\ & = p_1 \ end {align} $$
Wenn wir also über den Grundzustand des Partikels sprechen in einer Box mit der Länge $ L $, unabhängig davon, ob Sie die positive Wellenfunktion verwenden
$$ \ psi_1 = \ sqrt {\ frac {2} {L}} \ sin {\ left (\ frac { \ pi x} {L} \ right)} $$
oder die negative Wellenfunktion
$$ - \ psi_1 = - \ sqrt {\ frac {2} {L}} \ sin {\ left (\ frac {\ pi x} {L} \ right)} $$
oder die komplexe Wellenfunktion
$$ i \ psi_1 = i \ sqrt { \ frac {2} {L}} \ sin {\ left (\ frac {\ pi x} {L} \ right)} $$
Sie erhalten genau die gleichen Werte für die durchschnittliche Position $ (= L / 2) $, durchschnittlicher Impuls $ (= 0) $ und durchschnittliche Energie $ (= h ^ 2 / 2mL ^ 2) $ (das Wort Durchschnitt ist hier redundant, da dies ein stationärer Zustand ist, aber was auch immer)
Alles, was ich bisher gesagt habe, kann leicht auf drei Dimensionen verallgemeinert werden. Es kann auch auf lineare Kombinationen stationärer Zustände verallgemeinert werden, dh Lösungen der zeitabhängigen Schrödinger-Gleichung.
Eine Anmerkung zu Molekülorbitalen
"Okay, aber was passiert, wenn Sie Atomorbitale zu Molekülorbitalen kombinieren? Sie haben konstruktive Interferenzen durch die positive + positive und destruktive Interferenz durch die positive + negative, aber was ist mit der negativen + negativen Kombination?"
"Gute Frage, ich selbst!"
Lassen Sie uns über das $ \ ce {H2} $ -Molekül sprechen. Der richtige Weg, um die Molekülorbitale zu finden, besteht darin, die Schrödinger-Gleichung für das gesamte System zu lösen, was wirklich schwierig ist. Eine Möglichkeit, ungefähre Formen der MOs zu finden, besteht darin, lineare Kombinationen von Atomorbitalen zu erstellen. Diese Methode wird als LCAO-Näherung bezeichnet. Nennen wir das 1s-Orbital des Wasserstoffs links $ \ phi_1 $ und das 1s-Orbital des Wasserstoffs rechts $ \ phi_2 $. In den vorhergehenden Abschnitten haben wir bereits festgestellt, dass für das Wasserstoffatom die einzelnen Phasen von $ \ phi_1 $ und $ \ phi_2 $ keine Rolle spielen . Nehmen wir der Einfachheit halber an, dass ihre Phasen beide positiv sind.
Nach dem, was Sie bereits wissen, können Sie zwei Molekülorbitale $ \ psi_1 $ und $ \ psi_2 $ erhalten:
$$ \ begin {align} \ psi_1 & = \ phi_1 + \ phi_2 \\\ psi_2 & = \ phi_1 - \ phi_2 \ end {align} $$
Dies sind die bindenden und antibindenden Orbitale jeweils (zumindest innerhalb einer Normalisierungskonstante, die mir hier nicht wichtig ist, da die Details irrelevant sind). Lassen Sie uns nun über die Kombinationen sprechen, die wir verpasst haben.
$$ \ begin {align} - \ phi_1 - \ phi_2 & = - \ psi_1 \\ - \ phi_1 + \ phi_2 & = - \ psi_2 \ end {align} $$
Wir haben bereits gesagt, dass $ \ psi_1 $ und $ \ psi_2 $ (Annäherungen an) Lösungen für die Schrödinger-Gleichung sind. Das bedeutet, dass nach dem, worüber wir zuvor gesprochen haben, $ - \ psi_1 $ und $ - \ psi_2 $ auch (Annäherungen an) Lösungen für die Schrödinger-Gleichung sein müssen. Sie müssen die gleichen Energien wie $ \ psi_1 $ und $ \ psi_2 $ haben. Soweit das Molekül weiß (und sich darum kümmert), sind sie dasselbe wie $ \ psi_1 $ und $ \ psi_2 $.
Nun, seitdem Die einzelnen Phasen der Atomorbitale spielen keine Rolle. Wenn Sie wirklich möchten, können Sie der ganzen Welt erklären, dass Sie Folgendes definieren:
$$ \ phi_3 = \ phi_1 \ text {und} \ phi_4 = - \ phi_2 $$
d.h. Das linke Wasserstoff-1s-Orbital $ \ phi_3 $ ist positiv und das rechte Wasserstoff-1s-Orbital $ \ phi_4 $ ist negativ. In diesem Fall können Sie die Molekülorbitale konstruieren:
$$ \ begin {align} \ psi_1 & = \ phi_3 - \ phi_4 \\\ psi_2 & = \ phi_3 + \ phi_4 \ end {align} $$
Die Koeffizienten der Atomorbitale müssten unterschiedlich sein, da Sie darauf bestanden , sie in verschiedenen Phasen zu haben - jedoch Das Ergebnis ist das gleiche! Sie erhalten ein bindendes MO und ein antibindendes MO.